A pharmaceutical manufacturer’s decision to stay with their CDMO through commercialization or switch to a different partner is one of the most important decisions in a drug development program. It affects the timeline, regulatory risk, budget, and chances of a successful drug launch. This decision is often made later than ideal, under pressure, and with not enough information. Staying with the same CDMO is often the stronger strategic choice, but only when the right planning happens early and sponsors are honest about what continuity actually requires.
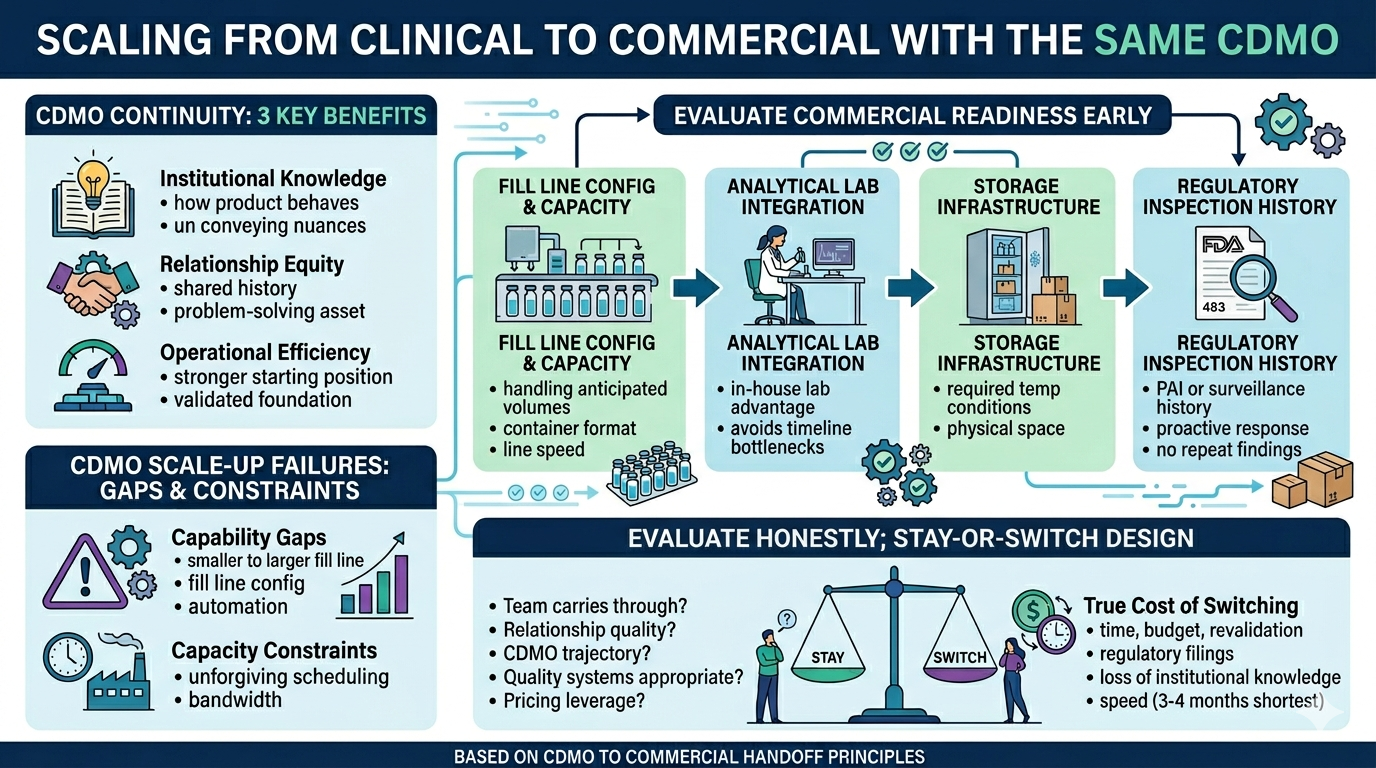
CDMO Continuity: 3 Key Benefits
Remaining with a clinical CDMO offers benefits across three dimensions:
- Institutional knowledge. A CDMO that has filled a product through clinical development knows things that no transfer package can fully convey: how the product behaves during formulation, what anomalies appeared and how they were resolved, and which approaches were tried before the current process was settled upon.
- Relationship equity. A team that has worked with a program from the start has a better understanding of a client’s priorities, risk tolerance, and communication style. This shared history helps with problem-solving on a commercial scale. In this setting, slow decisions are costly, and mistakes are more difficult to correct.
- Operational efficiency. A client choosing to scale with their current CDMO means beginning from a stronger position. The commercialization work still needs to happen, including equipment validations, PPQ batches, and stability programs. However, the work builds on an existing foundation instead of building from scratch.
CDMO Scale-Up Failures: Gaps & Constraints
There’s only a strong case for scaling with the same CDMO if they can support commercial manufacturing, which some cannot. This is often due to structural gaps rather than quality failures.
CDMO Capability Gaps
Capability gaps are the more common scale-up problem. While a CDMO may have run clean and reliable clinical batches, they might not have the fill line configuration, automation, or infrastructure required for commercial scale fill-finish.
CDMO Capacity Constraints
Capacity constraints are another potential issue. Commercial supply scheduling is unforgiving in ways that clinical scheduling is not. A CDMO may have the capability but not the bandwidth.
Both are avoidable with early due diligence, but clinical batch records alone will not surface them.
Evaluate CDMO Commercial Readiness Early
Commercial capability doesn’t reveal itself through clinical performance alone. Sponsors need to evaluate it directly and deliberately. The assessment should focus on four areas:
- Fill line configuration and capacity. Can the CDMO’s equipment handle the anticipated batch size, container format, fill volume, and line speed at commercial scale?
- Analytical lab integration. An integrated, in-house lab is a meaningful operational advantage at commercial scale. Release testing and stability programs scale with manufacturing volume, and if the lab is outsourced or under-resourced it becomes a timeline bottleneck regardless of fill line performance. Outsourced testing adds time and introduces dependency the sponsor doesn’t control.
- Storage infrastructure. Does the CDMO have physical space to store commercial batches at the product’s required temperature conditions?
- Regulatory inspection history. Has the site previously had a pre-approval inspection or a commercial surveillance inspection? A site that has never been inspected in a commercial setting is an unknown variable in your timeline. If the site has received 483 observations, ask how they were responded to. A thorough, proactive response with no repeat finding is a more meaningful signal than the existence of observations alone.
How to Evaluate the CDMO Stay-or-Switch Decision
Phase II is often a natural time to formalize this evaluation, when sponsors have considerable experience with their CDMO. They should evaluate honestly across five areas:
- Commercial capability and capacity. Three should be no questions left about fundamental capability questions.
- Relationship quality and issue history. How a CDMO handles setbacks is more revealing than how they perform when everything goes smoothly. Sponsors should think specifically about any issues that arose during clinical manufacturing and ask honestly: were they surfaced quickly, investigated thoroughly, and resolved in a way that built confidence? Sponsors should also ask whether the team that managed their clinical program will carry through to commercial. If the answer is no, the institutional knowledge and relationship equity built during clinical development doesn’t automatically transfer with the contract.
- The CDMO’s own trajectory. Is the CDMO expanding? What does that mean for a program — more capacity, or more competition for slots? A CDMO’s future matters as much as their present, because a commercial relationship extends years beyond approval.
- Quality systems and documentation. A quality system that works for clinical use might not be suitable for the scale needed for commercial supply. Sponsors should check if electronic batch records, LIMS, and document control platforms have audit trail features, access controls, and system validation.
- Inspection history. Inspection history matters too, but it requires careful reading. An FDA Form 483 is a list of observations, not a citation or warning letter. What matters is not whether a CDMO has ever received observations, but how they responded – whether findings were addressed at root cause, and whether the same issues recur across inspections. A CDMO with a mature quality culture will discuss their regulatory history openly and specifically. Evasiveness is a more meaningful red flag than the existence of observations in a public database.
- The true cost of switching. Account for what a technology transfer actually requires: time, budget, revalidation work, regulatory filings, and the loss of institutional knowledge the current CDMO holds about your product.
CDMO Tech Transfer: Real Costs & Timelines
Sponsors often underestimate their own burden in technology transfer – one of the most time and resource-intensive steps in a drug development program. When a sponsor moves to a new CDMO, this is a large undertaking regardless of organization, which requires engineering runs, method qualification across incoming, in-process, and release testing, new batch records, specification rewrites, change parts sourcing. Even with a straightforward program and a strong transfer package, the fastest technology transfer can realistically be completed is three to four months. Complex programs, incomplete documentation, or novel formats extend that timeline.
There is also knowledge that no transfer package captures. This includes the experimental history behind current process decisions and the practical know-how that experienced operators have about a product. This knowledge often does not make it into the batch record notes. When the same team manages a program from clinical to commercial, that knowledge remains within the team.
PPQ Timeline: Pre-Steps Before Validation
Treating commercial validation as a single event rather than a sequence of activities is a common mistake. Before PPQ can begin, significant steps must have already been taken:
- Media fills completed and any anomalies resolved
- Filter validation and extractables and leachables work completed for the specific product and filter combination
- Sterilization validation established for all equipment and product contact parts
- Engineering or pilot runs completed to resolve any open process questions at commercial scale
The PPQ process itself typically takes between eight and ten weeks, not including any of the pre-work listed above. Sponsors who plan according to the PPQ timeline without considering what comes before it will find themselves falling behind before the campaign begins.
Three cost drivers need close attention in any commercial validation budget: lab technology transfer, program complexity, and capital expenses for change parts or equipment specific to a product. A CDMO that identifies capital expense needs early provides sponsors with information they can plan around. One that brings them up late creates a problem for sponsors that they can only react to.
Negotiate CDMO Pricing Post-Approval
Once a product is commercially approved with a specific CDMO, switching costs are real. The questions sponsors should ask is whether that dynamic affects their commercial pricing leverage.
It can, if the CDMO is oriented toward short-term value extraction. But a CDMO whose business model depends on long-term relationships and repeat programs across a sponsor’s pipeline has a different incentive structure. Pricing a locked-in client aggressively is a short-term gain against a potential long-term loss, in a market where sponsor networks are tight and reputation travels. The stay-vs-switch evaluation is the right moment to ask how the CDMO approaches commercial contracting. A CDMO confident in their partnership model should answer that question directly.
Make the Decision Deliberately
The stay-or-switch decision is made well depending on one thing: whether it’s made deliberately and with the right information. Sponsors who evaluate commercial capability as part of their ongoing program management – rather than waiting until the decision is forced – have more information, more time, and more leverage over the outcome, whatever that outcome turns out to be.
Continuity is not always the right choice. Some CDMOs cannot support commercial manufacturing, and the right move is to find that out and act on it. Others can, and the relationship and institutional knowledge built through clinical development becomes a genuine asset at commercial scale. Neither conclusion is universal. The goal is simply to reach the decision with clear eyes.